Research Interests
Research Interests
Necroptosis is a form of newly defined programmed cell death. The fundamental characteristic of necroptotic cell death is the loss of membrane integrity and release of intracellular contents, the execution of which is dependent on the membrane-disrupting activity of MLKL after its phosphorylation by RIPK3. In recent decades, necroptosis cell-released components, also known as damage-associated molecular patterns (DAMPs), have attracted the most interest from researchers studying their pro-inflammatory activities. Excessive or uncontrolled necroptosis contributes to human diseases such as rheumatoid arthritis and neurological disorders. Pile of works on the pro-inflammatory role of necroptosis have established the detrimental side of necroptotic cell-released factors. However, whether necroptotic cell-released factors have any beneficial effect under physiological conditions is entirely unknown.
Combining mouse genetics with techniques of molecular biology, cell biology, and biochemistry, we are dedicated to investigating the ‘beneficial roles’ of necroptosis in the context of tissue regeneration, which has been largely neglected for long; and to deciphering the molecular regulations of necroptosis signaling that balances cell survival or aberrant cell division.
An ‘amyloidase’ reverses functional amyloid formation to prevent necroptosis
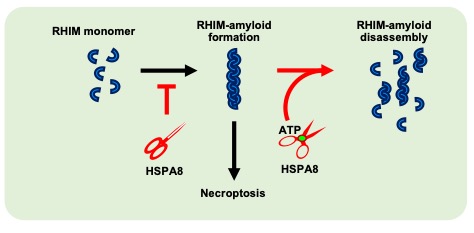
 Amyloids are distinguished by their ultra-stable fibrous structure. In contrast to canonical disease-related amyloids, emerging research indicates that a significant number of cellular amyloids, termed ‘functional amyloids’, contribute to signal transduction as temporal signaling hubs in humans. However, it is unclear how these functional amyloids are effectively disassembled to terminate signal transduction. RHIM motif-containing amyloids, the largest functional amyloid family discovered thus far, play an important role in mediating necroptosis signal transduction in mammalian cells. Recently, we identified heat shock protein family A member 8 (HSPA8) as a new type of enzyme — which we name as ‘amyloidase’ — that directly disassembles RHIM-amyloids to inhibit necroptosis signaling in cells and mice. The discovery that HSPA8 acts as an amyloidase dismantling functional amyloids provides a fundamental understanding of the reversibility nature of functional amyloids, a property distinguishing them from disease-related amyloids that are unbreakable in vivo.
Amyloids are distinguished by their ultra-stable fibrous structure. In contrast to canonical disease-related amyloids, emerging research indicates that a significant number of cellular amyloids, termed ‘functional amyloids’, contribute to signal transduction as temporal signaling hubs in humans. However, it is unclear how these functional amyloids are effectively disassembled to terminate signal transduction. RHIM motif-containing amyloids, the largest functional amyloid family discovered thus far, play an important role in mediating necroptosis signal transduction in mammalian cells. Recently, we identified heat shock protein family A member 8 (HSPA8) as a new type of enzyme — which we name as ‘amyloidase’ — that directly disassembles RHIM-amyloids to inhibit necroptosis signaling in cells and mice. The discovery that HSPA8 acts as an amyloidase dismantling functional amyloids provides a fundamental understanding of the reversibility nature of functional amyloids, a property distinguishing them from disease-related amyloids that are unbreakable in vivo.

The ‘altruistic role’ of necroptosis for tissue regeneration
Although excessive necroptosis causes inflammatory disorders, we speculate that under physiological conditions, when necroptosis is performed to a degree that does not exceed the organism's innate homeostatic capability, their true physiological purpose will become apparent. Utilizing a muscle regeneration model, we identified Tenascin-C (TNC), which is secreted by necroptotic myofibers, as a critical component that promotes muscle stem cell (MuSC) proliferation during regeneration by directly triggering EGF receptor (EGFR) cascade. This discovery unveiled a hitherto uncharacterized “altruistic physiological role” of necroptosis. In the meantime, the complexity of necroptotic cell-released substances and the entire process of tissue regeneration inspired us to further investigate the numerous roles of the ‘necroptotic niche’ in tissue repair.


Checkpoint of necroptotic membrane-damaging
The hallmark of necroptosis is Mixed Lineage Kinase Domain-Like protein (MLKL)-mediated membrane damage, which leads to cellular contents release. The gold standard in clinical research and the benchmark of testing necroptosis has been designated as the activation of MLKL, specifically the phosphorylation of its C-terminal at T357/S358 by Receptor Interacting Protein Kinase 3 (RIPK3). Surprisingly, a substantial number of phospho-MLKL-containing cells can survive necroptosis in all necroptosis-sensitive cell lines. By collecting these ‘survivor cells’, we recently identified an inhibitory phosphorylation modification on the N-terminal MLKL that blocked necroptosis execution. Moreover, the phospho-dead mutant MlklS82G/S82G knock-in mice showed spontaneous necroptosis, which led to multi-organ inflammation, and even infant mortality. This newly identified phosphorylation on MLKL act as a checkpoint to prevent cytolytic necroptosis. This phosphorylation regulation on MLKL is potentially a therapeutic target of necroptosis over activation-induced inflammatory injury.


Future Directions:
1. Necroptosis-mediated trans-tissue communication
2. Necroptosis sensitivity control
3. Functional amyloid regulation
4. MLKL-mediated membrane rupture